
Les amyloses cardiaques regroupent trois maladies dont le processus physiopathologique commun est caractérisé par une accumulation extracellulaire de protéines fibrillaires insolubles infiltrant la matrice extracellulaire cardiaque principalement au niveau du myocarde (mais aussi de l’endocarde et du péricarde). L’accumulation progressive de fibrilles amyloïdes se traduit par une augmentation de l’épaisseur et de la rigidité du cœur conduisant à une insuffisance cardiaque sévère initialement avec une conservation de la fraction d’éjection ventriculaire gauche (IC à FEVG préservée). Le tissu nerveux cardiaque peut également être atteint comme le système nerveux autonome. D’autres organes peuvent être infiltrés en fonction du type d’amylose : rein, nerf, foie, langue, tube digestif… pouvant entraînés des présentations cliniques complexes et hétérogènes brouillant les pistes et expliquant souvent un retard diagnostic de plusieurs mois ou années.
La classification des amyloses repose sur la nature biochimique de la protéine amyloïde impliquée dans la formation des dépôts. Ces fibrilles étant constituées de nos propres protéines, elles sont non immunogènes et le corps humain se laisse tranquillement infiltré sans aucune réaction du système inflammatoire. Les amyloses cardiaques les plus fréquentes sont les amyloses AL (immunoglobuliniques, la mieux connue liée à une production anormale de chaînes légères kappa ou lambda), les amyloses à transthyrétine (TTR) : héréditaire (TTR mutée) liées à une mutation dans le gène de la transthyrétine et les amyloses séniles (TTR sauvage, dont le mécanisme n’est pas encore élucidé).
De la clinique au diagnostic
Les manifestations cardiaques sont non spécifiques pouvant comporter des symptômes d’insuffisance cardiaque et/ou des troubles conductifs et rythmiques. Les manifestations extra-cardiaques sont diverses et varient en fonction du type d’amylose : syndrome du canal carpien, ecchymose périorbitaire, macroglossie, etc. L’atteinte nerveuse périphérique se traduit cliniquement par des troubles sensitifs associés à des paresthésies des extrémités. Il n’est pas rare que les symptômes neurologiques soient attribués à tort à d’autres causes neurologiques notamment chez le sujet âgé (neuropathie diabétique, canal lombaire étroit, spondylodysthésis, etc.). L’atteinte du système nerveux autonome peut être au premier plan (forme AL et forme TTR héréditaire) et toucher toutes les fonctions autonomes entraînant des hypotensions orthostatiques sévères, des gastroparésies responsables de vomissements incoercibles sources de dyskaliémie, des troubles des fonctions génito-urinaires.
Le diagnostic d’amylose au cabinet en cas de forme cardiaque repose sur : un interrogatoire sur les antécédents familiaux et chirurgicaux (canal carpien), un examen clinique complet pour mettre en évidence les atteintes d’organes, l’ECG à la recherche d’un microvoltage, une échocardiographie pour objectiver l’infiltration myocardique et ses conséquences sur l’hémodynamique cardiaque (diminution de la déformation myocardique basale).
À l’issue de la consultation, la prescription d’un bilan sanguin complet, la recherche d’une gammapathie monoclonale à chaîne légère comportant une électrophorèse sérique, une immunofixation, un dosage de chaîne légère libre et la protéinurie de Bence Jones associées à la prescription d’une IRM cardiaque permettra bien souvent d’apporter des éléments diagnostics. L’IRM mettra en évidence l’infiltration myocardique.
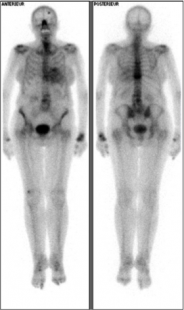
La scintigraphie cardiaque aux diphosphonates complétera le bilan par la fixation cardiaque du traceur osseux, phénomène non compris mais très utile. Les amyloses à transthyrétine sont associées à une forte fixation du traceur au niveau cardiaque. À l’inverse les amyloses cardiaques AL entraîne peu ou pas de fixation. La poursuite du bilan devra se réaliser dans des centres ayant l’expertise génétique et anatomopathologiques.
Une nouvelle ère thérapeutique !
La prise en charge cardiaque des amyloses nécessite de traiter la cause (le processus physiopathologique) et ses conséquences cardiologiques. Le traitement cardiologique diffère totalement de l’insuffisance cardiaque. Arrêt des bêtabloquants, discussion sur le bénéfice des IEC. Prévention des accidents emboliques et des troubles conductifs et rythmiques doivent faire systématiquement discuter le traitement anticoagulant ou la pose de pacemaker/défibrillateur. Les recommandations européennes de l’insuffisance cardiaque ne peuvent donc pas être utilisées pour traiter ces patients.
Le traitement des amyloses AL a beaucoup évolué et l’utilisation de nouvelles chimiothérapies (Bortezomid, dexaméthasone…) a permis d’améliorer considérablement le pronostic des patients. La réponse à la chimiothérapie s’associe à une normalisation des symptômes à la diminution des biomarqueurs et des chaînes légères.
Le traitement médical des amyloses à transthyrétine est en pleine évolution. Historiquement, le traitement des amyloses héréditaires consiste à réaliser une greffe hépatique qui doit être, en cas d’atteinte cardiaque importante, combinée avec une greffe cardiaque. La transplantation hépatique vise à supprimer la source de production de trasnthyrétine mutée, synthétisée dans le foie, en la remplaçant par un foie synthétisant une transthyrétine sauvage. De nouveaux traitements médicaux sont apparus avec des propriétés pharmacologiques nouvelles et sont testés dans de nombreux essais thérapeutiques. Ces traitements vont du blocage de la production de la protéine transthyrétine par des ARN messagers anti-sens ou des oligonucléotides, la stabilisation de la protéine rendue instable par la mutation génétique, ou la production d’anticorps spécifiques de la forme fibrillaire de la transthyrétine ou de la substance amyloïde P composant tous les types de fibrilles amyloïdes. Ces derniers traitements permettraient de traiter les formes les plus tardives de la maladie car conduisant à l’activation du système immunitaire et au retrait des dépôts d’amyloses intramyocardiques.
Ces nouveaux traitements sont en cours d’évaluation et feront probablement partie de l’arsenal thérapeutique de l’insuffisance cardiaque. L’ère des thérapies ciblées en cardiologie a débuté !
Réseau Amylose Mondor - centre de référence national - amyloses cardiaques (filière CARDIOGEN, centre de référence cardiomyopathies et trouble du rythme héréditaires et rares). Service de cardiologie CHU Henri-Mondor et GRC Amyloid Research Institute (Créteil)
www.reseau-amylose-chu-mondor.org
(1) González-López E et al. Wild-type transthyretin amyloidosis as a cause of heart failure with preserved ejection fraction. Eur Heart J 2015 Oct 7;36(38):2585-94
(2) Damy Tet al. Prevalence and clinical phenotype of hereditary transthyretin amyloid cardiomyopathy in patients with increased left ventricular wall thickness. Eur Heart J 2016 Jun 14;37(23):1826-34
CCAM technique : des trous dans la raquette des revalorisations
Dr Patrick Gasser (Avenir Spé) : « Mon but n’est pas de m’opposer à mes collègues médecins généralistes »
Congrès de la SNFMI 2024 : la médecine interne à la loupe
La nouvelle convention médicale publiée au Journal officiel, le G à 30 euros le 22 décembre 2024